AI+math|从Alphafold说起
AI+math|从Alphafold说起
引言
作者:@同济大学 刘越
Github ID:@miracle-techlink
联系邮箱:miracle.techlink@gmail.com
校内邮箱: 2254018@tongji.edu.cn
特别感谢以下组织和个人提供的帮助:
Datawhale开源学习社区 (点击链接进入其github教程仓库)
集智俱乐部(https://pattern.swarma.org/user/116904) (点击链接进入官网)
@同济大学数学科学学院陈小杨教授 (邮箱:xychen100@tongji.edu.cn)
2024年诺贝尔化学奖授予大卫·贝克(David Baker)、戴米斯·哈萨比斯(Demis Hassabis)和约翰·江珀(John M.Jumper),以表彰他们在蛋白质设计和蛋白质结构预测领域作出的贡献。他们与Deepmind公司在2020年发布的lphaFold2算法,在蛋白质结构预测领域取得了重大突破,其预测结果与实验结果高度一致,为生物科学领域带来了革命性的影响。我们今天就一起走进AlphaFold的世界,了解其背后的原理,体验其强大的预测能力,并探讨分子结构预测问题在AI领域的学术前沿。

这次我们的分享主要分为三个部分:
- alphafold原理简介与demo体验
- AI+化学算法比赛解读与baseline体验
- 分子结构预测问题学术前沿探讨
参考链接:(如有侵权,请联系删除)
- AlphaFold的原理及解读
- 实用教程:使用AlphaFold2进行蛋白质结构在线预测
- Datawhale AI+化学算法比赛飞书教程
- 集智俱乐部讲座:计算蛋白质设计:从AlphaFold到强化学习
- 集智俱乐部讲座: 拓扑几何增强化学分子模型
Alphafold原理简介与demo体验
Alphafold 简介
蛋白质通过卷曲折叠会构成三维结构,蛋白质的功能正由其结构决定。了解蛋白质结构有助于开发治疗疾病的药物。而蛋白质的结构非常复杂,通过实验手段获取蛋白质结构非常困难,因此需要通过计算手段预测蛋白质结构。
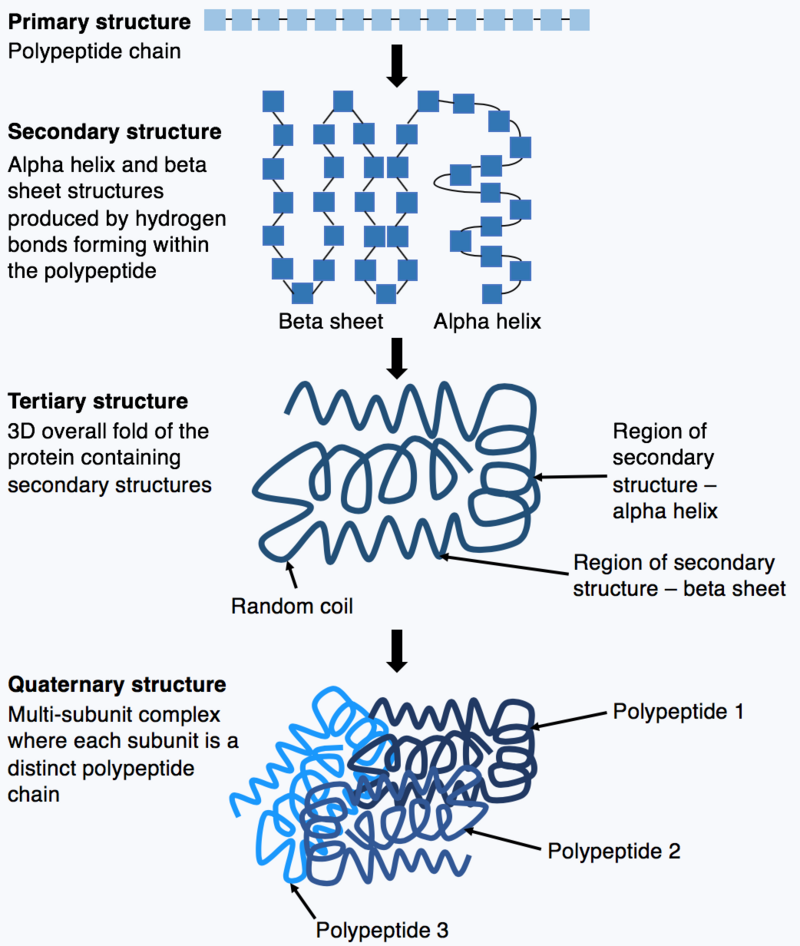
AlphaFold2是DeepMind公司于2020年发布的蛋白质结构预测算法,其核心思想是通过深度学习模型预测蛋白质的三维结构。AlphaFold2在蛋白质结构预测领域取得了重大突破,其预测结果与实验结果高度一致,为生物科学领域带来了革命性的影响。
AlphaFold2的主要创新点在于其使用了Transformer模型,这是一种基于自注意力机制的深度学习模型,可以有效地捕捉蛋白质序列中的长距离依赖关系。此外,AlphaFold2还使用了大量的蛋白质结构数据,通过大规模的训练,使得模型能够更好地预测蛋白质的三维结构。
Alphafold2 算法原理
Nature论文:Highly accurate protein structure prediction with AlphaFold
AlphaFold根据深度学习技术,通过输入蛋白质一级结构来解析二级结构及三级结构,而一级结构为一组蛋白质氨基酸序列,但是AlphaFold的特征并不只是输入蛋白质氨基酸序列,而包含了多种相关信息。
- 氨基酸的排列:AlphaFold会用一种特殊的编码方式(one-hot向量)来表示每个氨基酸,并记录序列的长度。
- 氨基酸的编号:它会为序列中的每个氨基酸分配一个编号。
- 相似蛋白质的比较:AlphaFold会寻找其他相似的蛋白质序列,来帮助预测当前蛋白质的结构。
- 不同蛋白质的比较:除了相似的,它也会看看那些不相似的蛋白质序列。
- 氨基酸之间的相互作用:AlphaFold还会考虑氨基酸之间的位置关系和角度关系。
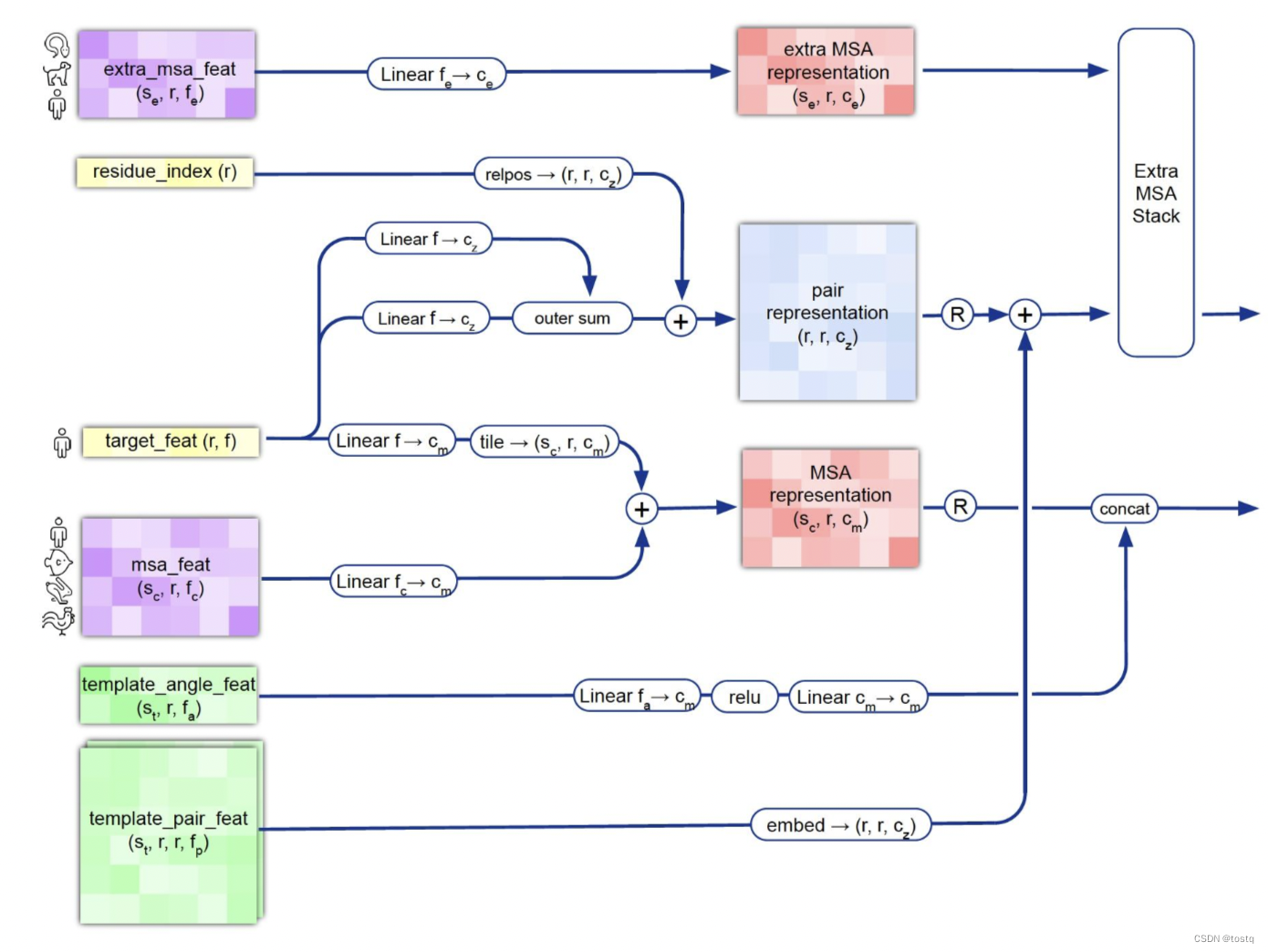
所有这些信息会被处理和整合,然后输入到AlphaFold的神经网络中。这个网络有几个重要的部分:
AlphaFold 的架构包含多个关键组件,以下是其主要组成部分的总结:
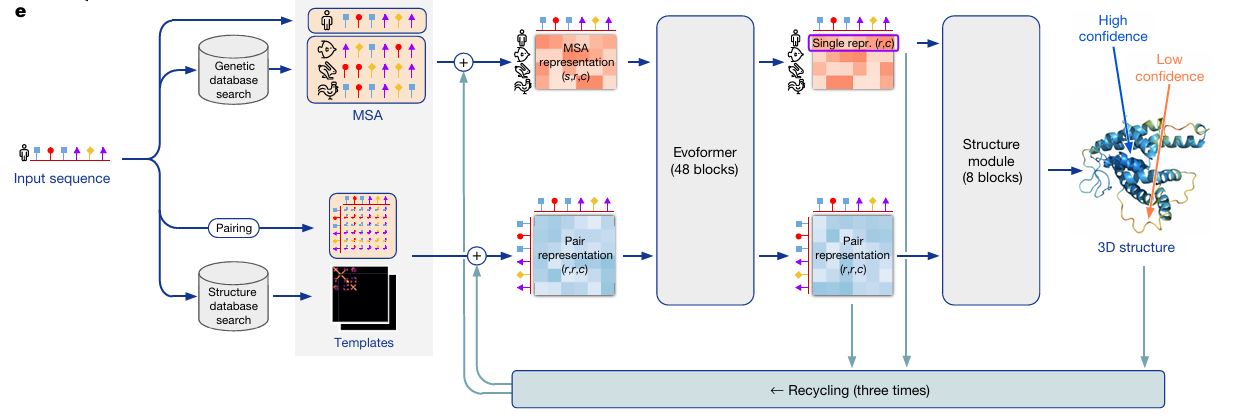
- 多序列比对:帮助AlphaFold理解蛋白质序列的进化关系。
- Evoformer模块:处理输入的信息,并提取重要的特征。
- 结构模块:开始构建蛋白质的三维模型。
- 注意力机制:让模型能够关注到序列中重要的部分。
- 迭代细化:通过多次迭代来提高预测的准确性。
- 端到端预测:直接从氨基酸序列预测出三维结构。
- 自我蒸馏和数据增强:通过预测未标记的序列来提高模型的准确性。
- 损失函数和评估指标:使用多种方法来评估和优化模型的预测。
Alphafold2 demo体验
若需要用源项目复现的同学,可以参考以下链接:
AlphaFold2源码
alphafold2_jax预测蛋白质三维结构的深度学习模型
对于大多数做生物的同学,其实并不关心AlphaFold2的原理,也没有使用服务器的条件,只想简单方便地预测一个蛋白质的结构。那谷歌的Colaboratory也提供了在线使用版本。如果没有梯子的话,也没有关系,可以在讲座结束后自行下载体验。
请同学们点击以下链接,体验一下AlphaFold2的强大功能:AlphaFlod Colab
进入Colab界面后,只需要输入自己想预测的序列和项目名称,即可:
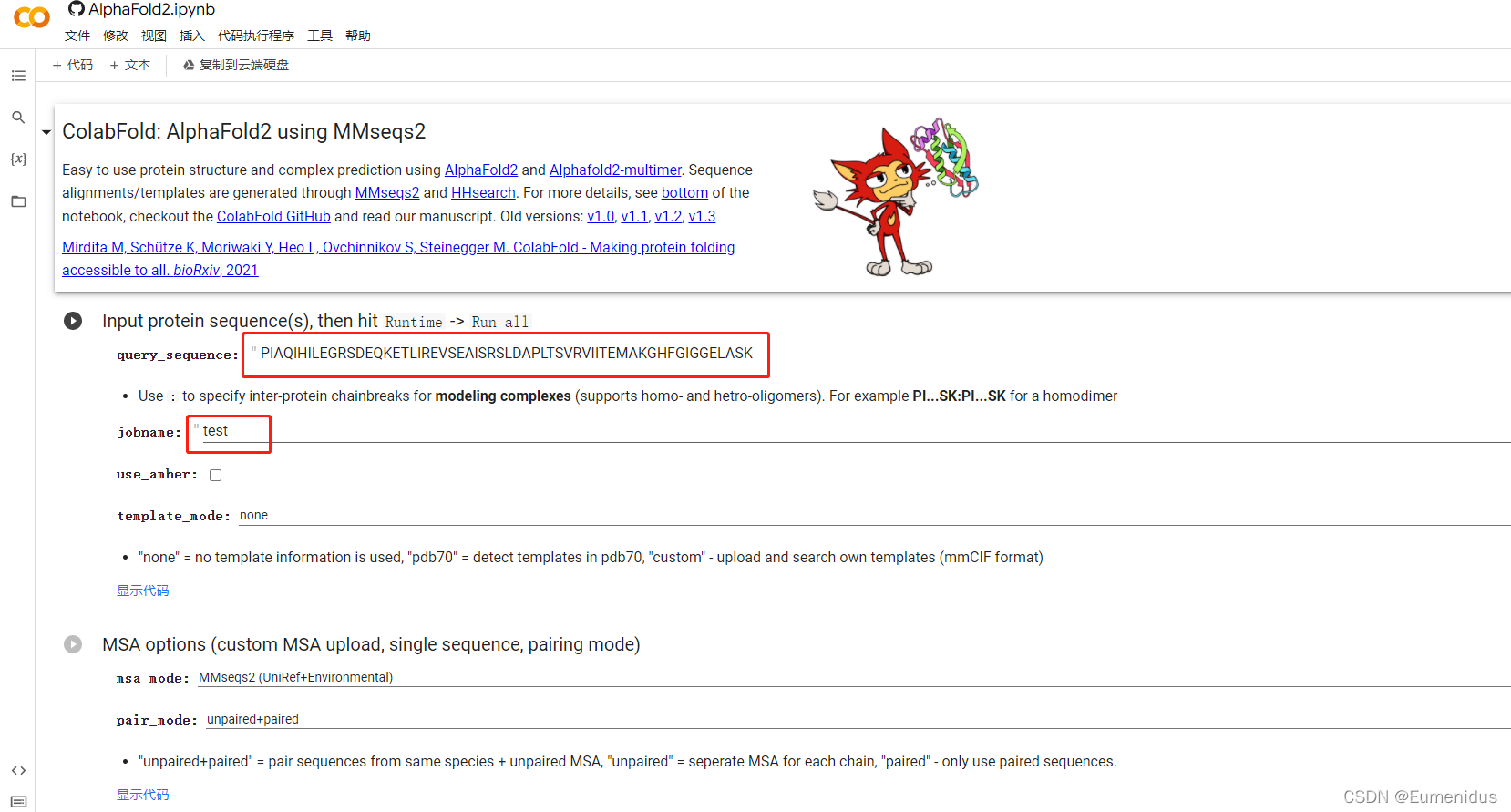
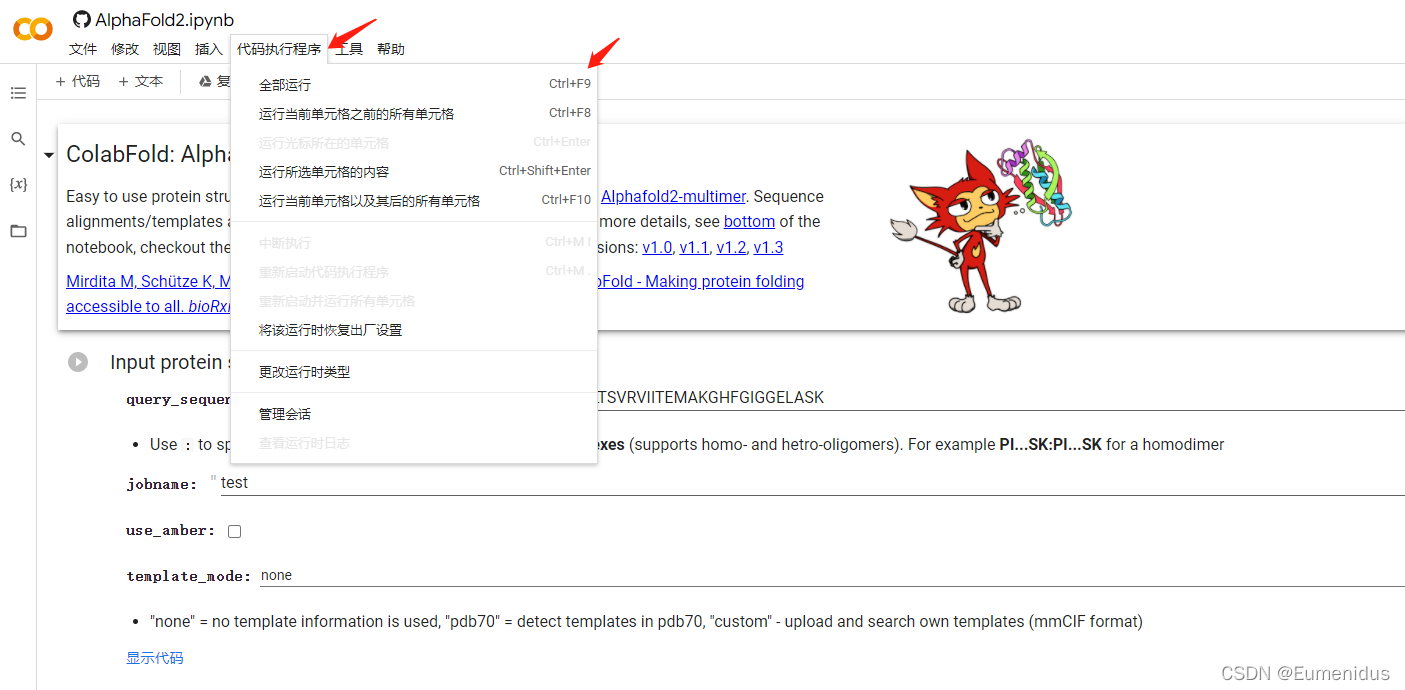
运行方法也很简单,选择代码执行程序->全部运行即可
AlphaFold2参数修改
除了使用默认设定参数外,还可以根据实际需求修改部分参数,如:Advanced settings中的number_recycles,这个参数可以理解为是每个模型的循环次数,默认是3,循环次数越多相对应的准确度也会高一点,但运行时间也会越长。
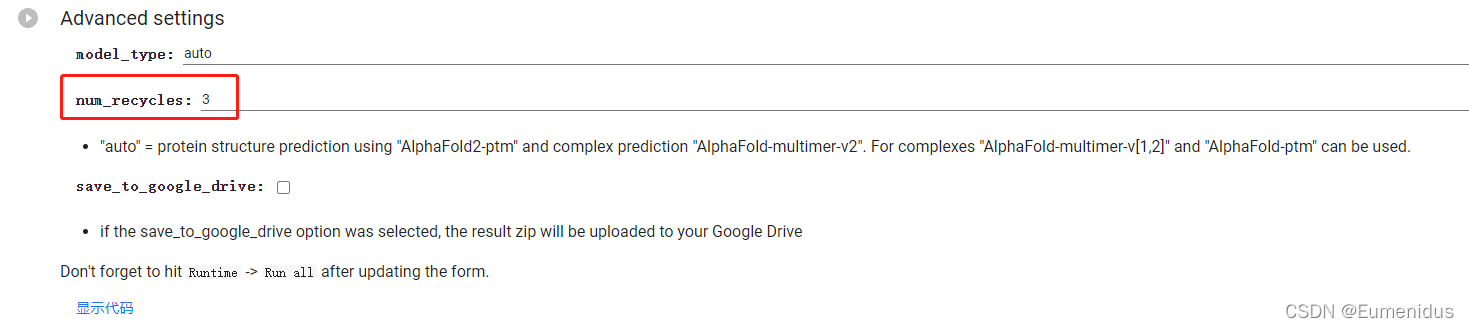
每次运行所产生的模型数也可以修改,在Run Prediction中,点击显示代码即可展开代码片:
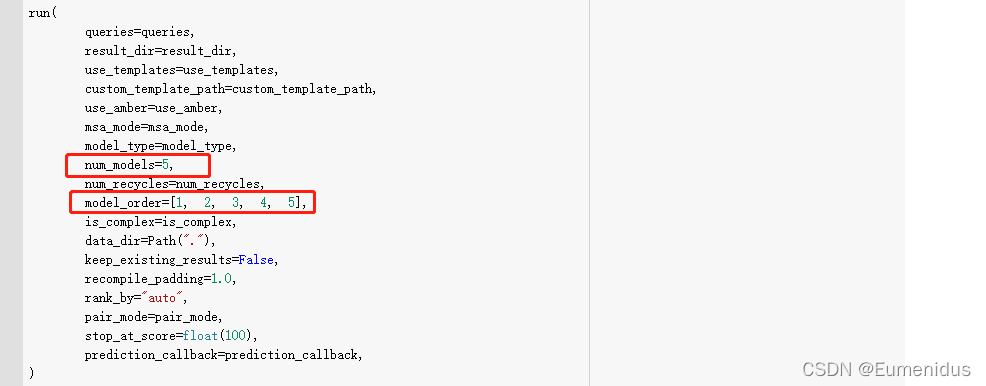
其中我们要修改的是num_moldels,直接改数字即可修改每次运行时产生的模型数。如果模型数大于5的话,最好把下面的model_order也跟着修改了,就把数字补齐到你的模型数即可。
耐心等到运行结束,就可以看到预测的蛋白质结构了,点击3D View即可查看:
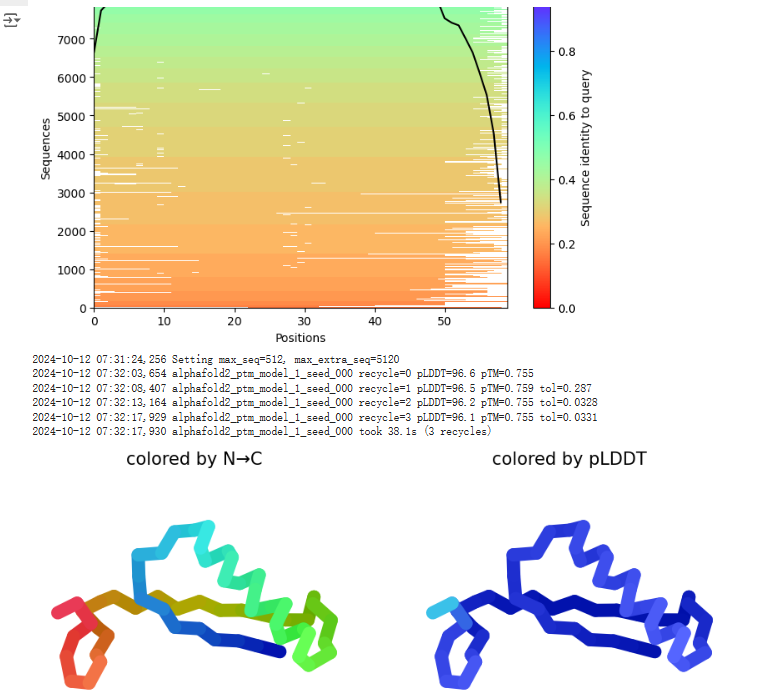
AI+化学算法比赛解读与baseline体验
赛题解读
背景知识
碳氮成键反应、Diels-Alder环加成反应等一系列催化合成反应,被广泛应用于各类药物的生产合成中。研究人员与产业界在针对特定反应类型开发新的催化合成方法时,往往追求以高产率获得目标产物,也即开发高活性的催化反应体系,以提升原子经济性,减少资源的浪费与环境污染。然而,开发具有高活性的催化反应体系通常需要对包括催化剂和溶剂在内的多种反应条件进行详尽的探索,这导致了它成为了一项极为耗时且资源密集的任务。这要求对包括催化剂和溶剂在内的多种反应条件进行详尽的探索。目前,反应条件的筛选在很大程度上依赖于经验判断和偶然发现,导致催化反应条件的优化过程既耗时又费力,并且严重制约了新的高效催化合成策略的开发。
反应底物和反应条件是决定其产率的关键因素。因此,我们可以利用AI模型来捕捉底物、条件与产率之间的内在联系。借助产率预测AI模型,仅需输入底物和条件的信息,我们就能够预测该反应组合下的产率,从而有效提升催化反应的条件筛选效率。
任务目标
- 构建一个能够准确预测碳氮成键反应产率的预测模型
通过对反应中所包含的反应底物、添加剂、溶剂以及产物进行合理的特征化,运用机器学习模型或者深度学习模型拟合预测反应的产率,或者利用训练集数据对开源大语言模型进行微调以预测反应的产率。
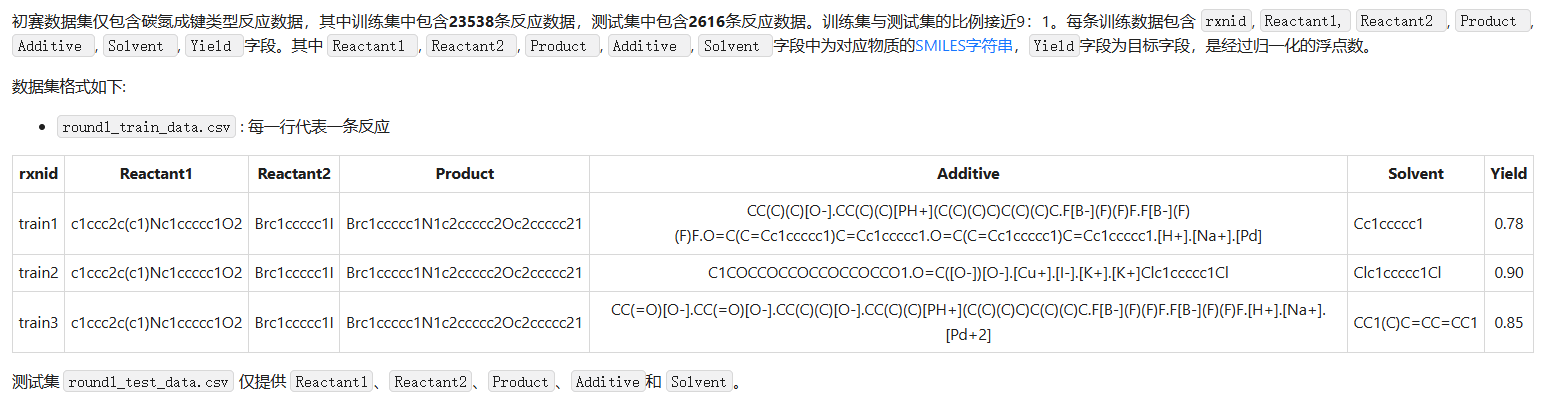
数据集介绍
评价指标
Baseline:基于SMILES与随机森林的化学分子结构预测
首先,我们打开Datawhale的飞书教程—教程链接,并且找到baseline1的代码压缩包,将其下载到本地。
随后我们在网页端打开modelscope的Notebook页面(没有注册账号的同学可以用手机号注册一个),进入其中的CPU云环境。
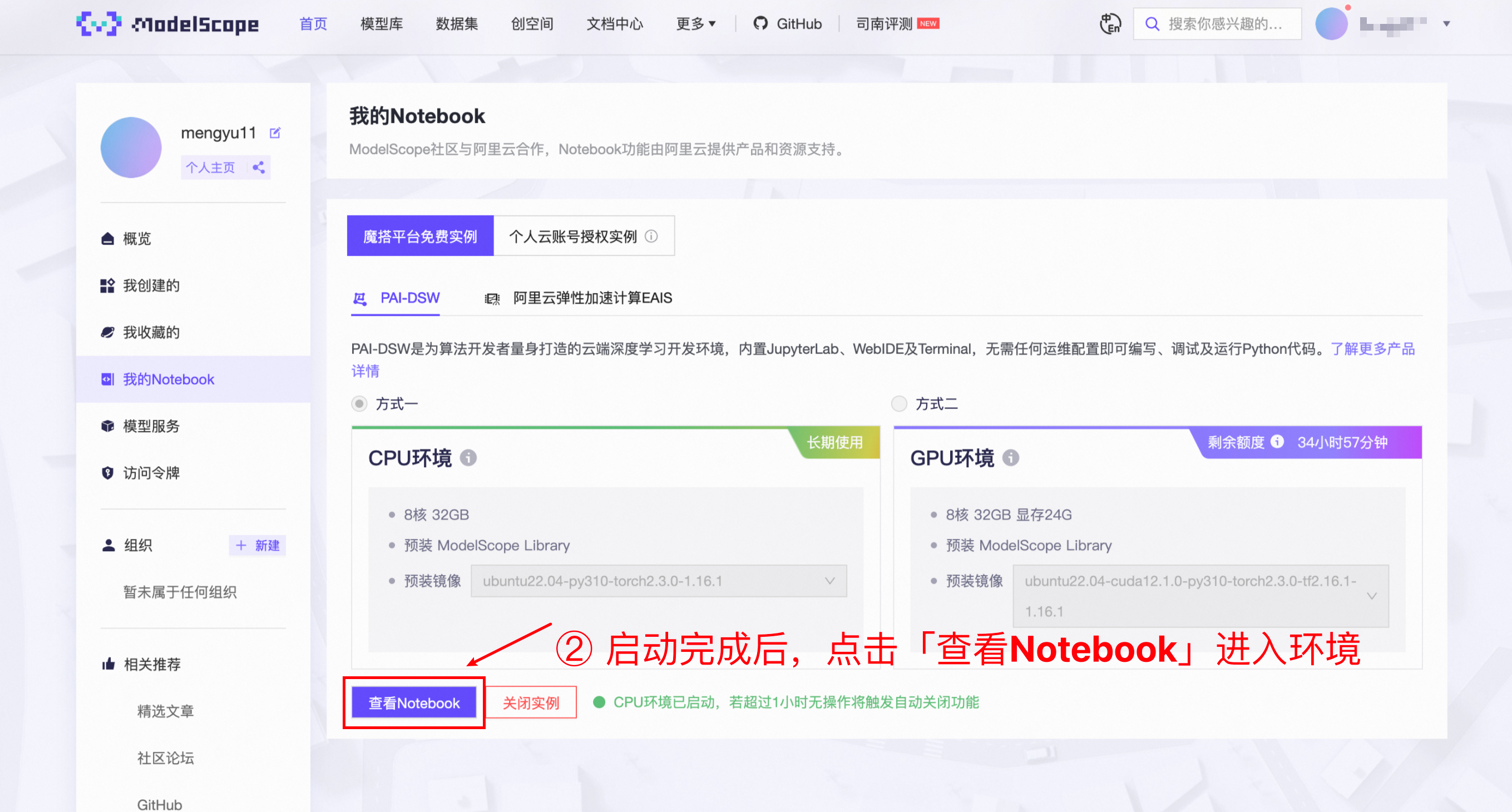
在Notebook中,我们需要完成以下操作:
1. 将下载好的代码压缩包上传到云环境中。
2. 打开终端terminal。
3. 在终端输入 unzip AI+化学baseline文件包.zip ,解压文件。
4. 进入mp文件夹,进入code二级目录,找到Task1_baseline.ipynb文件。
5. 重启kernel,点击 “run all” 运行代码。
6. 执行完成后,下载submit.txt,查看预测结果。
接下来,乘着代码运行的时间,我们来分析一下baseline的代码,看看它到底是怎么实现的。
特征表示方法
无论是蛋白质分子结构,还是化学分子,键的位置信息。如何采用合适的特征表示方法,将分子结构信息转化为模型可读的输入,都是长时间以来研究者们关注的重点。例如,一开始例如,科学家们尝试通过一些规则来提取特征,并将其保存在数据库中。但是,这种方法存在一些问题,比如,它不能很好地处理分子中的环结构,而且特征提取过程比较复杂。
SMILES —— 最流行的将分子表示为序列类型数据的方法
SMILES,提出者Weininger et al[1],全称是Simplified Molecular Input Line Entry System,是一种将化学分子用ASCII字符表示的方法,在化学信息学领域有着举足轻重的作用。当前对于分子和化学式的储存形式,几乎都是由SMILES(或者它的一些手足兄弟)完成的。使用非常广泛的分子/反应数据库,例如ZINC[2],ChemBL[3],USPTO[4]等,都是采用这种形式存储。SMILES将化学分子中涉及的原子、键、电荷等信息,用对应的ASCII字符表示;环、侧链等化学结构信息,用特定的书写规范表达。以此,几乎所有的分子都可以用特定的SMILES表示,且SMILES的表示还算比较直观。
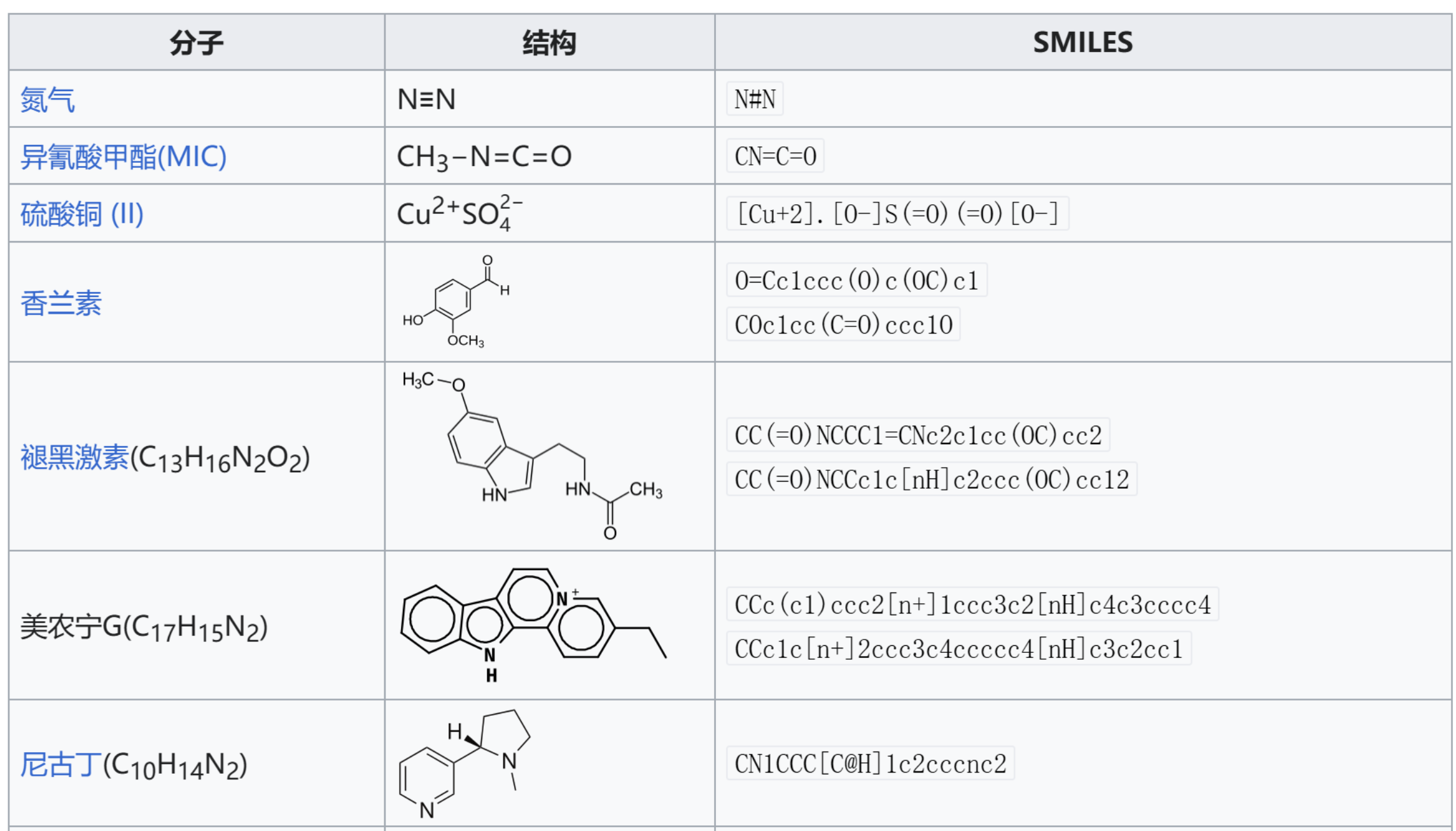
官方发布的数据是对化学分子的SMILES表达式,具体来说,有rxnid,Reactant1,Reactant2,Product,Additive,Solvent,Yield字段。其中:
- rxnid 对数据的id标识,无实际意义
- Reactant1 反应物1
- Reactant2 反应物2
- Product 产物
- Additive 添加剂(包括催化剂catalyst等辅助反应物合成但是不对产物贡献原子的部分)
- Solvent 溶剂
- Yield 产率 其中Reactant1,Reactant2,Product,Additive,Solvent都是由SMILES表示。
1 | def mfgen(mol,nBits=2048, radius=2): |
分子指纹 —— 分子向量化
分子的指纹就像人的指纹一样,用于表示特定的分子。分子指纹是一个具有固定长度的位向量(即由0,1组成),其中,每个为1的值表示这个分子具有某些特定的化学结构。例如,对于一个只有长度为2的分子指纹,我们可以设定第一个值表示分子是否有甲基,第二个位置的值表示分子是都有苯环,那么[0,1]的分子指纹表示的就是一个有苯环而没有甲基的分子。通常,分子指纹的维度都是上千的,也即记录了上千个子结构是否出现在分子中。

我们可以用python中的RDKIT库来计算分子指纹。RDkit是化学信息学中主要的工具,是开源的。网址:http://www.rdkit.org,支持WIN\MAC\Linux,可以被python、Java、C调用。几乎所有的与化学信息学相关的内容都可以在上面找到。常用的功能包括但不限于:
- 读和写分子;
- 循环获取分子中原子、键、环的信息;
- 修饰分子;
- 获取分子指纹;
- 计算分子相似性;
- 将分子绘制为图片;
- 子结构匹配和搜索;
- 生成和优化3D结构。
1 | # 从csv中读取数据 |
在预测算法中,我们选取机器学习中经典的随机森林进行建模。
sklearn (scikit-learn)
这是一个非常广泛使用的开源机器学习库,基于Python,建立在NumPy、SciPy、Pandas和Matplotlib等数据处理和分析的库之上。它涵盖了几乎所有主流机器学习算法,包括分类、回归、聚类、降维等。API设计亲民,整个使用简单易上手,非常适合作为机器学习入门的工具。 官网:https://scikit-learn.org/stable/index.html
在sklearn中,几乎所有的机器学习的流程是:
1.实例化模型(并指定重要参数);
2.model.fit(x, y) 训练模型;
3.随机森林
参数解释:
1.n_estimators=10: 决策树的个数,越多越好;但是越多意味着计算开销越大;
2.max_depth: (default=None)设置树的最大深度,默认为None;
3.min_samples_split: 根据属性划分节点时,最少的样本数;
4.min_samples_leaf: 叶子节点最少的样本数;
5.n_jobs=1: 并行job个数,-1表示使用所有cpu进行并行计算
1 | # Model fitting |
Baseline优化思路:基于transformer架构或大模型
虽然传统的机器学习模型在化学分子预测任务中取得了显著的成果,但它们通常需要大量的特征工程和复杂的模型结构来捕捉分子中的复杂关系。近年来,基于Transformer的模型在自然语言处理和计算机视觉等领域取得了巨大的成功,并在化学分子预测任务中也得到了广泛的应用,其相比机器学习的优点如下:
1.XGBoost和随机森林等传统模型虽然也能捕捉非线性关系,但它们通常需要更多的特征工程来提取有用的信息,并且对于数据中长距离依赖的捕捉能力较弱。而且需要将序列数据转换为固定长度的特征向量,这可能会丢失一些重要信息。
2.化学分子可以通过SMILES字符串来表示,这是一种序列数据。Transformer模型,特别是序列到序列(Seq2Seq)模型,非常擅长处理序列数据。它们可以捕捉到序列中长距离的依赖关系,这对于理解分子中原子和键的关系至关重要。
3.而Transformer模型核心—注意力机制,它允许模型在处理序列时动态地关注序列的不同部分。在化学反应中,某些原子或键可能对产率有更大的影响,注意力机制可以帮助模型识别这些重要的特征。
4.在计算策略上,Transformer模型可以并行处理整个序列,这对于处理大规模的化学分子数据非常有用。
具体可以参考以下教程内容:基于transformer架构的化学反应预测 , Baseline链接
当然,也可以结合当下最火爆的大模型技术来进行研究,我们可以通过提示词工程(Prompt Engineering)来引导大模型进行预测,结合GPT-3、ChatGLM等模型。
1 | 你精通预测药物合成反应的产率,你的任务是根据给定的反应数据准确预测反应的产率(Yield)。请仔细阅读以下说明并严格遵循: |
具体可以参考教程:大模型预测飞书教程
分子结构预测问题学术前沿探讨
无论是化学分子结构预测还是蛋白质结构预测,都是人工智能领域的重要研究方向。近年来,随着深度学习技术的快速发展,越来越多的研究者开始关注这些问题的解决方法。在本文中,我们将介绍一些分子结构预测问题的学术前沿,并探讨如何使用深度学习技术来解决这些问题。
拓扑几何与分子结构预测
研究背景
在材料科学研究中,准确预测材料的性质对于新材料的开发和现有材料的性能改进至关重要。机器学习(ML)技术能够通过学习大量的数据构建预测模型,从而加速材料设计过程。在这一过程中,描述符或特征的选择对于机器学习模型的性能有着决定性的影响。传统的密度泛函理论(DFT)计算虽然准确,但是计算成本高且耗时。因此,研究者们一直在寻找一种既保持尺度不变性(旋转、平移不变)又能高效描述晶体化合物的方法。
该论文在nature上的链接:Topological representations of crystalline compounds for the machine-learning prediction of materials properties,
研究方法
本研究提出了一种基于原子特异性持续同调(ASPH)的方法,用于生成晶体化合物的拓扑表示,以预测它们的物理性质。研究方法包括以下几个关键步骤:

1.单形和单纯复形:定义了k-单形的概念,并构建了单纯复形来表示晶体结构。
2.同调:利用同调理论来描述晶体结构中的拓扑特征,如孤立点、环和空洞。
3.过滤和持续同调:通过改变过滤参数生成一系列拓扑空间,以捕捉不同尺度上的拓扑特征。
4.原子特异性持续同调:引入ASPH方法,通过考虑原子类型特异性,生成能够描述原子对和多体相互作用的拓扑特征。
5.拓扑表征:结合组成特征、化学计量属性、电子结构属性等,生成晶体化合物的拓扑表征。

研究者们应用了来自ICSD和OQMD的大型数据集,通过交叉验证将所提出的方法与Voronoi镶嵌和库伦矩阵等先前方法进行比较。
研究结果
研究结果表明,ASPH方法能够以较低的平均绝对误差(61 meV/atom)预测晶体化合物的性质。与Voronoi镶嵌和库伦矩阵等传统方法相比,ASPH方法在预测精度上有显著提升。此外,研究还分析了模型预测中误差较大的异常值与DFT计算的关系,并探索了部分化合物更容易导致模型预测产生较大偏差的原因。
ASPH方法的拓扑表征能够捕获成对和多体相互作用,并揭示一组原子在不同尺度上的拓扑性质关系。这种方法不仅提高了模型的性能,而且由于其基于代数拓扑的特性,提供了一种物理上更可解释的预测结果。
总结与展望
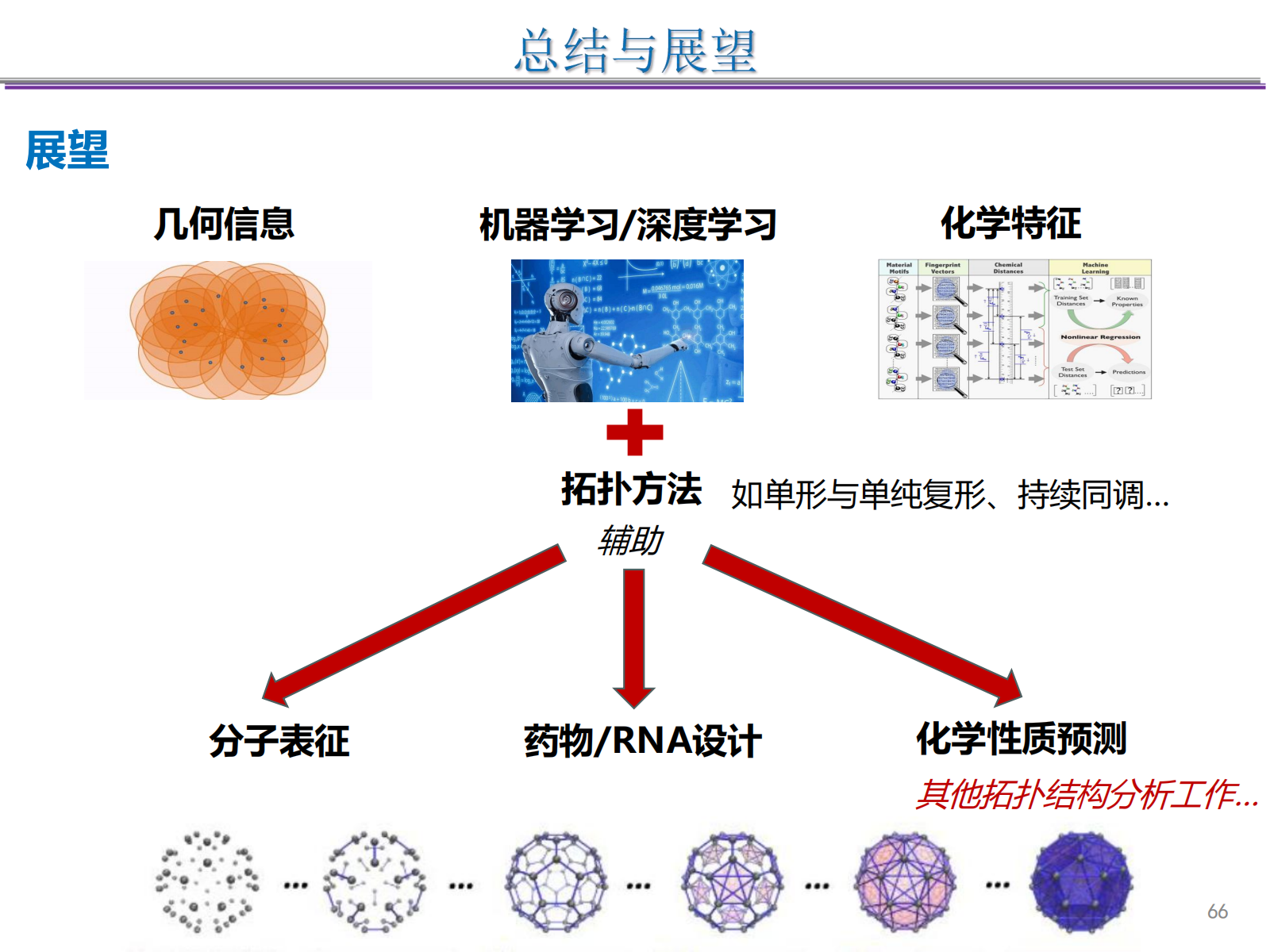
本研究成功地将ASPH方法应用于材料科学分析,并将多尺度几何信息嵌入拓扑不变量中,处理具有结构周期性和元素多样性的晶体化合物。尽管ASPH方法在小数据量下可能性能较差,且对低频元素的化合物预测能力有限,但它为发现具有理想性质的新材料提供了一种更快、更经济的方式。
未来的工作可以进一步优化ASPH方法,提高其对不稳定化合物和立体异构体的预测能力,并探索其在药物设计、RNA设计等领域的应用潜力。此外,拓扑方法还可以辅助其他拓扑结构分析工作,如单形与单纯复形、持续同调等,为化学性质预测提供新的视角。
研究汇报PPT文件:拓扑几何与分子结构
图神经网络与分子结构预测
研究背景
在化学信息学和材料科学中,分子结构的准确表示对于理解其化学性质和预测其行为至关重要。传统的分子表示方法,如SMILES字符串和基于图的方法,虽然在一定程度上能够描述分子的结构,但它们往往缺乏对分子几何特征和空间信息的捕捉。这种局限性在涉及立体化学和分子构象的任务中尤为明显。因此,研究者们一直在探索新的方法来更全面地表示分子结构,包括其几何和拓扑信息。
研究方法
本研究提出了一种基于离散里奇曲率的图感知嵌入方法,用于捕捉分子图中的几何特性和结构信息。这种方法的核心在于利用图的离散里奇曲率来描述分子图中的几何特征,而无需改变底层的神经网络架构。具体方法如下:
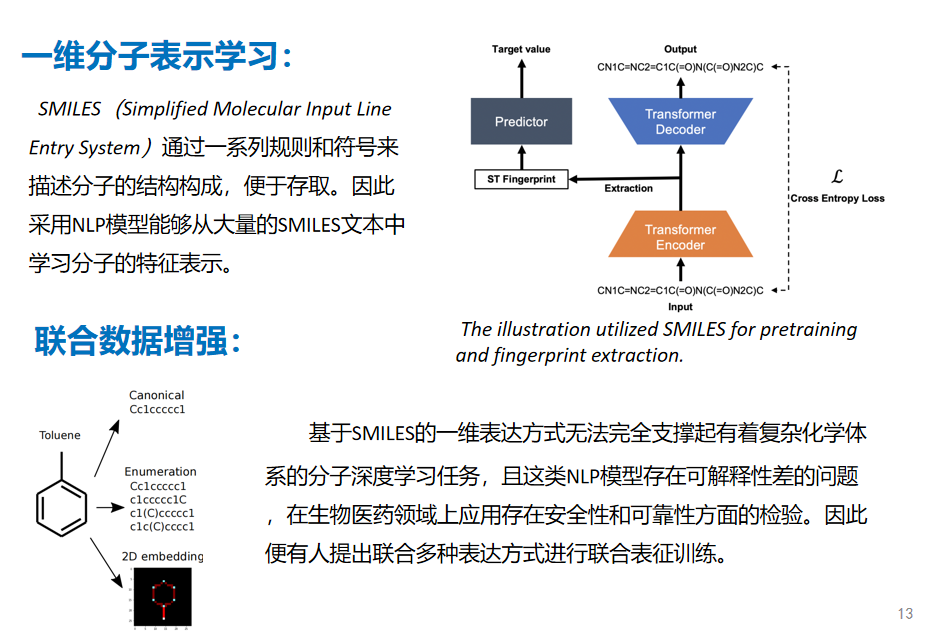
1.一维分子表示学习:使用SMILES字符串通过NLP模型学习分子的特征表示。
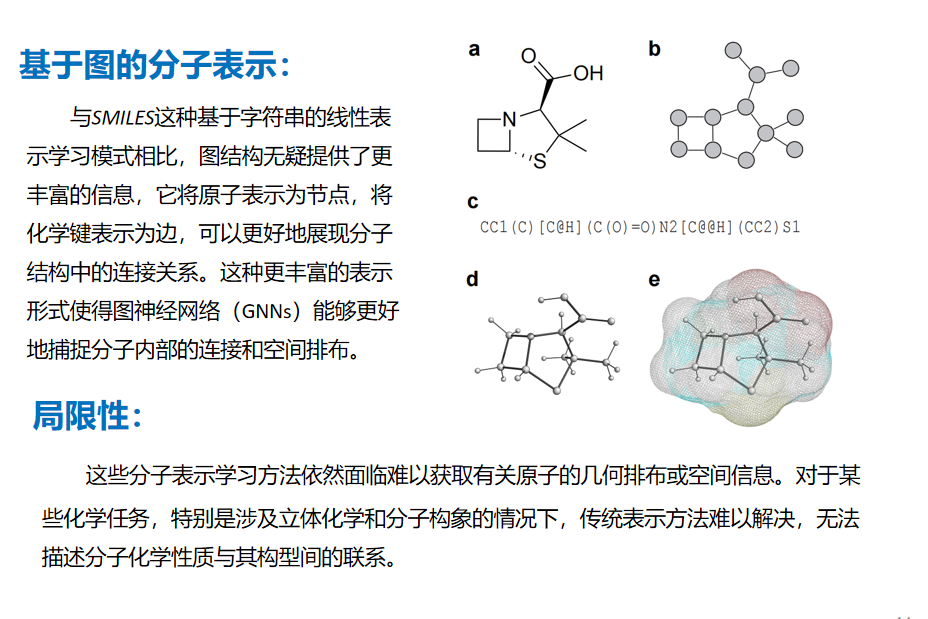
2.基于图的分子表示:将原子表示为节点,化学键表示为边,利用图神经网络(GNNs)捕捉分子内部的连接和空间排布。
3.节点特征编码:将化学信息编码进节点特征向量中,如原子序数、价态、电子层数、杂化类型等。
4.二维图数据增强:将图论信息编码进节点的特征向量中,如介数中心性、度中心性、图的拉普拉斯矩阵等。
5.三维图数据表示学习:使用等变图神经网络(GNNs)处理三维图数据,保持对三维空间欧几里得变换的等变性。
6.图的离散里奇曲率:计算分子图中各边的离散里奇曲率,捕捉分子图中的几何特性和结构信息。
7.基于曲率编码的分子图嵌入:将k-跳曲率编码嵌入到节点特征中,增强分子化学性质与几何拓扑信息之间的联系。
研究结果
研究结果表明,基于曲率的图感知嵌入能够有效地捕捉分子图中的几何特征和结构信息。这种方法能够区分具有相同度数但在不同分子中具有不同结构环境的原子。通过在k-跳子图上计算曲率值,研究者们能够扩展原始离散里奇曲率的定义,从而更好地揭示分子的几何特征与其化学性质之间的关系。
此外,将曲率编码集成到现有的图神经网络框架中,如GCN和Graphormer,可以显著提升模型对分子化学性质的预测能力。这种方法不仅提高了模型的性能,而且由于其无需对网络架构进行修改,因此具有很好的通用性和易集成性。
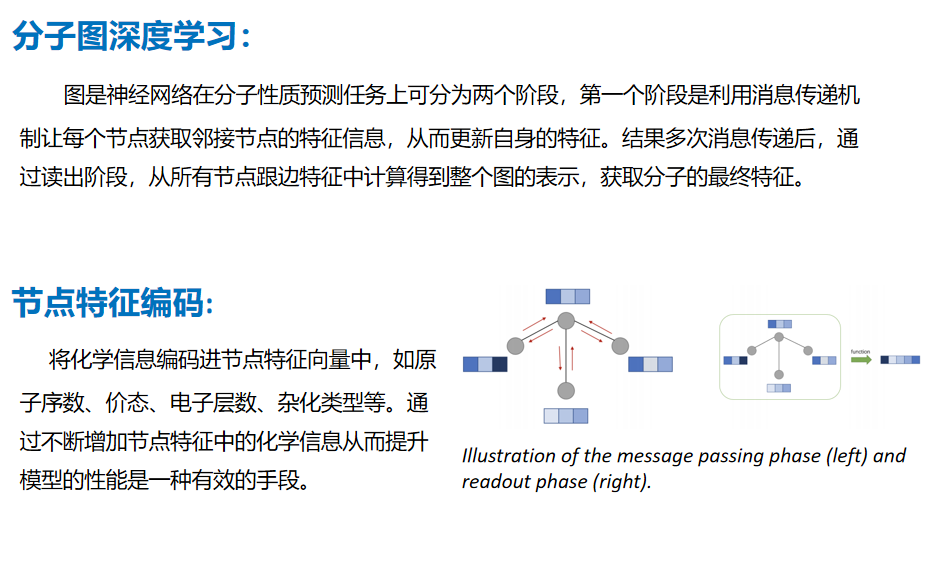
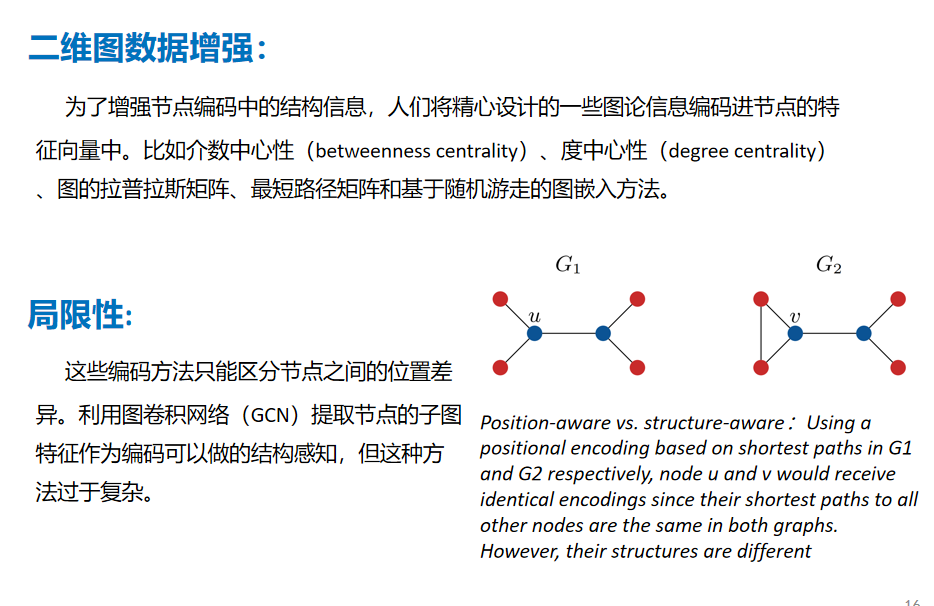
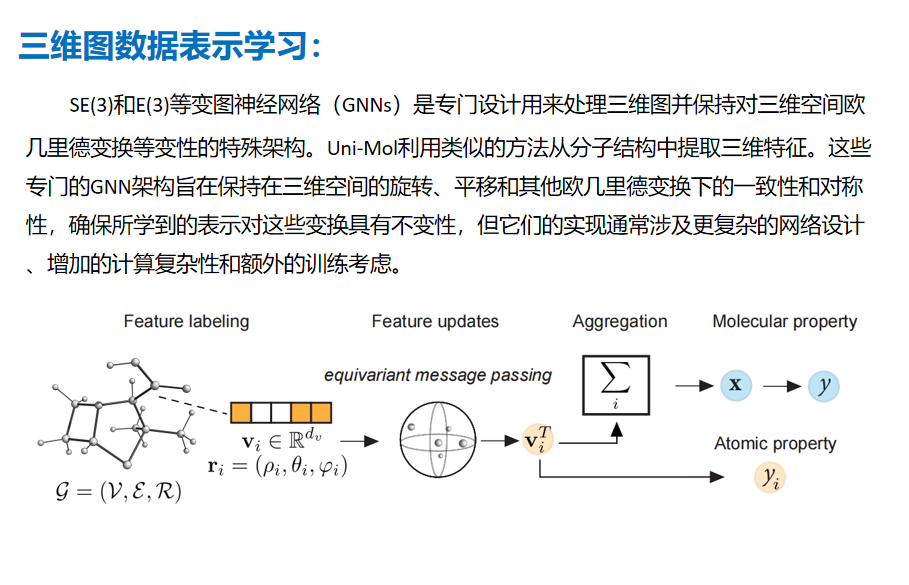
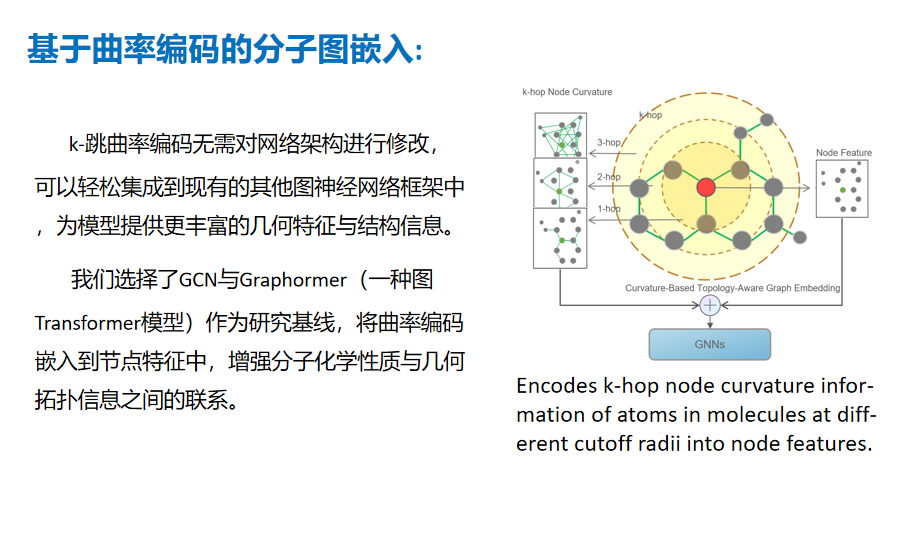
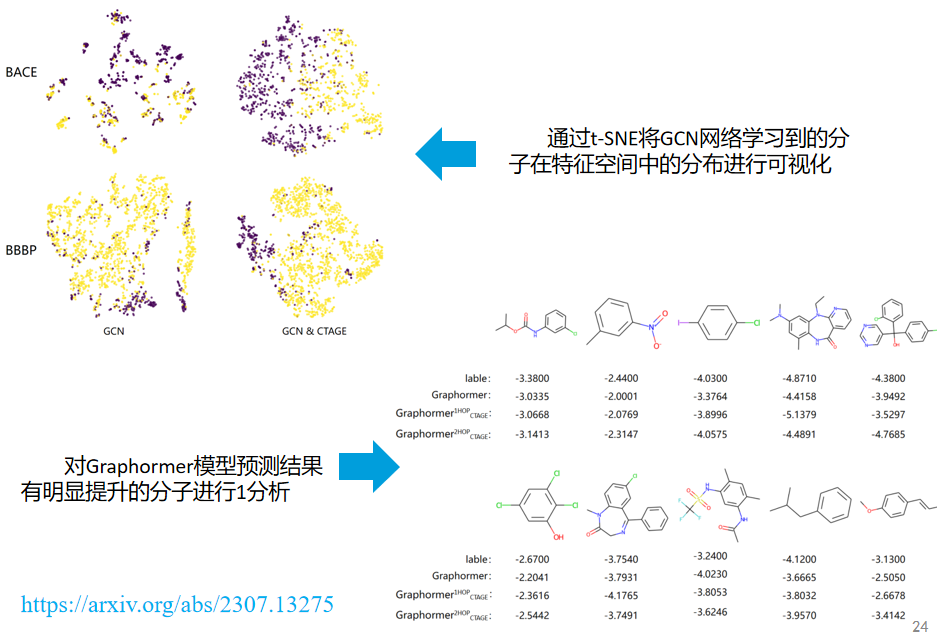
强化学习+蒙特卡洛树搜索与分子结构预测
引言
强化学习(RL)和蒙特卡洛树搜索(MCTS)是两种强大的机器学习方法,它们在许多领域都取得了显著的成果。比如著名的AlphaGo和AlphaZero,它们分别利用了强化学习和MCTS来在围棋游戏中取得胜利。这一算法主要应用在决策过程复杂、状态空间大、奖励延迟等问题上。在分子结构预测领域,RL和MCTS可以用于优化分子结构,以实现特定的化学性质或生物活性。
这一方法在今年数学建模国赛的B题中也展露头角,在工厂多工序,多流水线的决策问题中,RL+MCTS的方法可以有效解决决策搜索问题。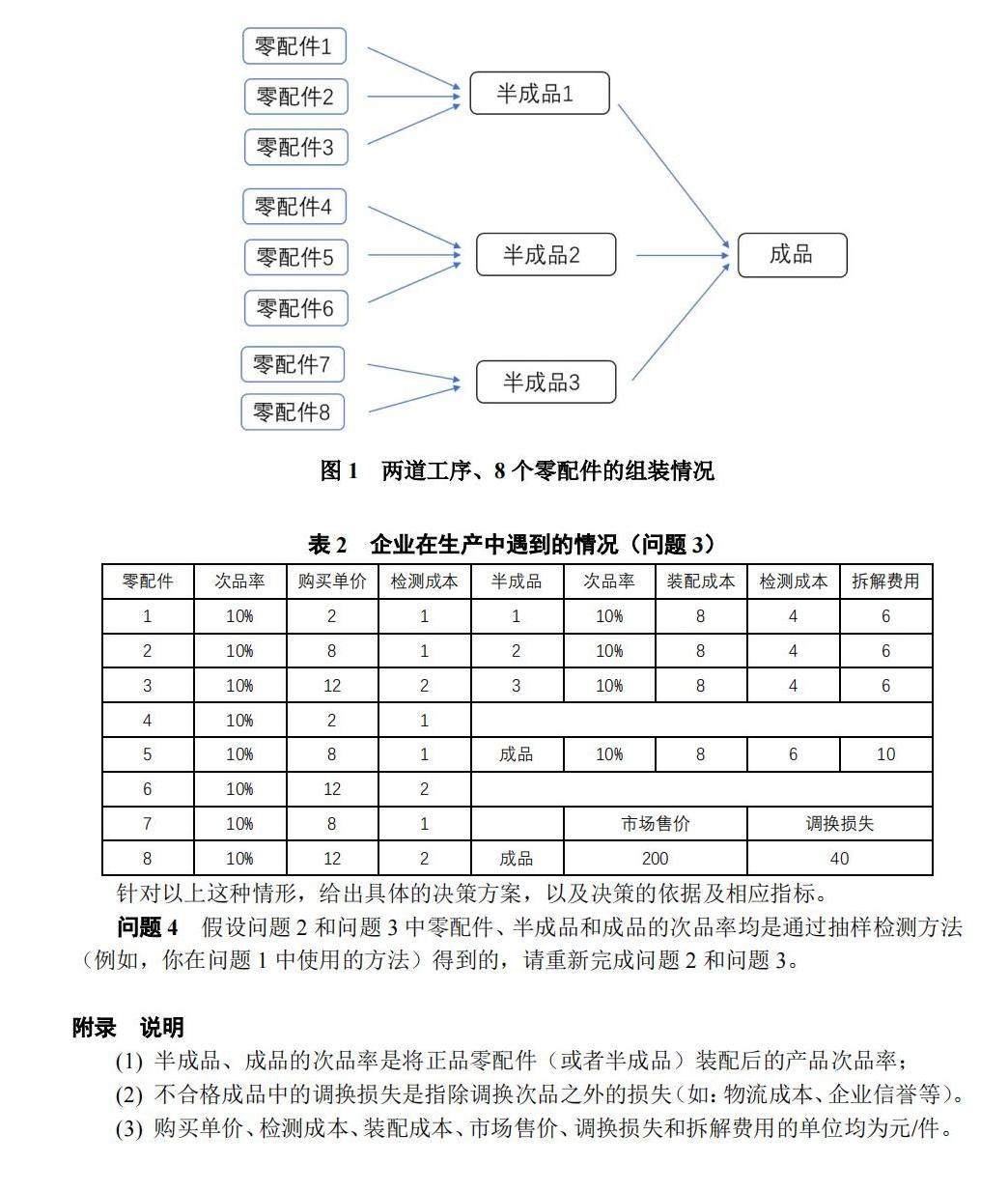
研究背景
自然进化过程中,多亚基蛋白质复合体的形成往往具有高度的形状互补性,以实现特定的生物学功能。然而,现有的蛋白质设计方法,特别是自下而上的层次化方法,通常无法实现这种通过进化优化的蛋白质结构。这些方法首先设计单体蛋白质结构,然后将它们组装成具有特定对称性的复合体,如二十面体或立方体结构。尽管这种方法在生物医学领域已有应用,如新设计的COVID疫苗,但它在设计复杂蛋白质纳米材料时存在局限性。
文章science链接:Top-down design of protein architectures with
reinforcement learning
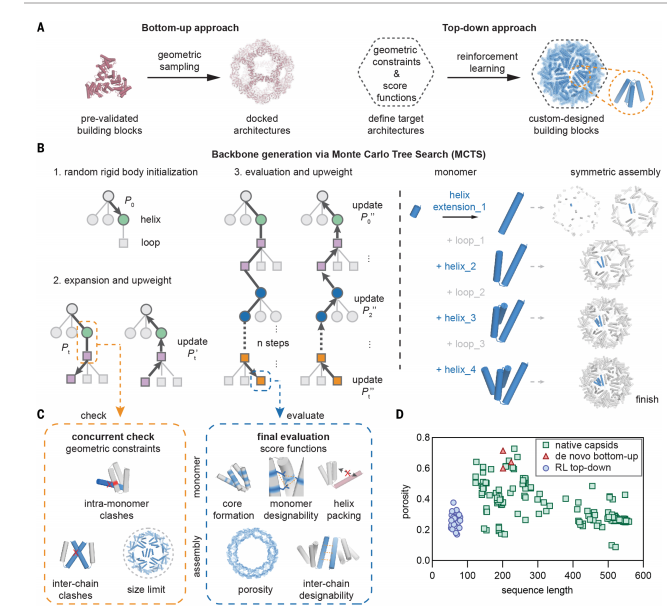
研究方法
为了克服这些局限性,研究者们开发了一种自上而下的蛋白质设计方法,该方法从所需的结构属性(如整体对称性、孔隙性等)出发,系统地构建能够优化这些属性的蛋白质亚基。这种方法的核心是使用蒙特卡洛树搜索(MCTS)算法,这是一种强化学习(RL)技术,用于在搜索树中找到最优的一系列选择。
MCTS算法实现步骤:
- 初始化:搜索树的构建从选择一个初始的螺旋片段开始,然后在树的每一步中,通过在N端或C端添加短蛋白质片段(螺旋或环)来扩展蛋白质链。
- 选择和扩展:在每个分支点,随机选择一个片段来扩展蛋白质链。选择是基于概率的,这些概率随着搜索的进行而更新。
- 几何约束:在搜索树的每一步中,应用几何约束来确保添加的片段不会违反预定的结构要求(如内部冲突和整体形状约束)。
- 评分函数:完成的蛋白质骨架使用评分函数进行评估,这些函数评估生成的结构如何满足用户指定的问题解决方案。
- 后向传播:根据评分函数的结果,重新调整搜索树中每个分支点的概率,以便后续迭代更有可能找到最优路径。
- 迭代优化:通过多次迭代搜索树,生成具有越来越高评分的完整蛋白质骨架。
- 多目标优化:通过初始化多个独立树并限制任何单一移动的最大概率来平衡探索与利用,从而优化全局结构属性。
蛋白质设计实现:
- 单体蛋白质设计:研究者们首先在单体蛋白质水平上测试了MCTS方法,选择生成具有任意预指定整体形状的蛋白质骨架作为测试问题。
- 对称纳米材料设计:将MCTS应用于设计具有所需对称性的纳米材料,如C5至C12的循环组装,以及多达240个亚基的二十面体、立方体和准对称二十面体组装。
- 纳米孔构建:研究者们将MCTS应用于填充两个先前设计的环形蛋白质之间的空间,以生成具有中心纳米孔的盘状结构。
- 迷你二十面体设计:研究者们探索了使用MCTS生成二十面体组装的方法,开发了特定的几何约束和评分函数来设计非常小且紧密包装的壳体。
研究结果
设计的盘状纳米孔和超紧凑型二十面体的冷冻电镜结构与计算模型非常接近。
设计的二十面体能够在疫苗反应和血管生成诱导中展示极高的免疫原和信号分子密度。
该方法能够设计具有所需系统属性的复杂蛋白质纳米材料,并展示了强化学习在蛋白质设计中的强大潜力。
学习资料推荐
最后给大家推荐两本很好的学习深度学习的书籍,尤其适合数学科学学院的同学。
1.math for AI.pdf
链接: https://pan.baidu.com/s/1y3-acj2XQq1Z4UBg0QjRHw 提取码: 27kv
2.Mastering PyTorch:Create and deploy deep learning models from CNNs to multimodal models, LLMs and beyond, 2nd Edition-Packt Publishing (2024).pdf
链接:https://pan.xunlei.com/s/VO8oBq5uQi23j4ADfX1wwS6BA1?pwd=g79d#